An Interview With A Mother of Two Children with Cystic Fibrosis
Home » Lifestyle »May is Cystic Fibrosis Awareness Month, and I take a photo a day of something purple as part of my Project 365 to help raise awareness of the disease. I also donate to help towards a cure, in the hope that one day CF will stand for Cure Found.
I have written in the past about why I donate to this cause, but today I’m bringing you an interview with the mother of two children with Cystic Fibrosis (CF). She is a dear friend, but we have never met in real life, because she lives on the other side of the world! One day though… Her two sweet little girls are who I donate for, and try to raise awareness for. When I see the snippets of her life through the photographs and videos she shares of the girls, well, I’m often left in awe of her strength as mother, and the fighting spirit that she has within her…
If you only read one post on my blog, please, make it this one…
Tell me about your family…
My name is Elisa and my husband’s name is Christopher. We live in Southern California, born and raised.
We have two wonderful daughters named Gwendolynn (6) and Genevieve (3). They are both so full of life.
They don’t let Cystic Fibrosis stand in their way, even if they aren’t feeling all that great.
They really do inspire us.


What is Cystic Fibrosis?
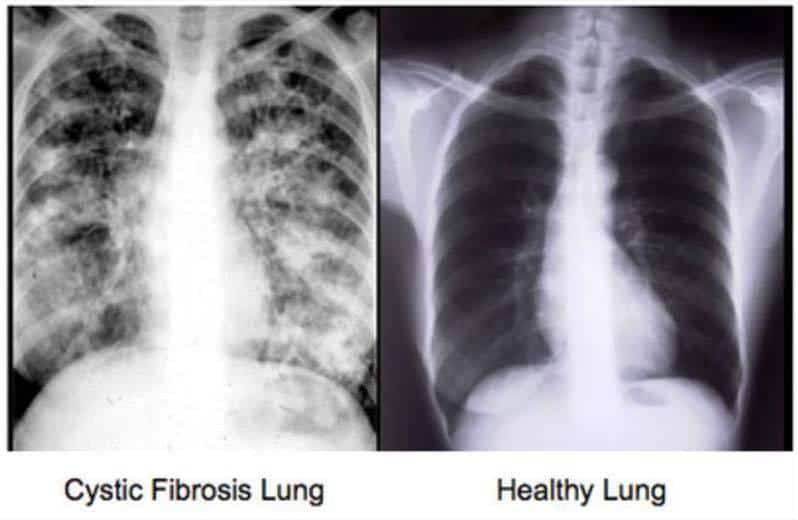
Cystic Fibrosis is a genetic life threatening disease. Both parents must carry a Cystic Fibrosis gene and every pregnancy they share is a 25% chance to have a child with CF. 1 in 25 people are carriers.
Cystic Fibrosis affects the cells that produce mucus, sweat and digestive juices. These secreted fluids are normally thin and slippery. But in Cystic Fibrosis, they are thick and sticky. Instead of acting as a lubricant, the secretions plug up tubes, ducts and passageways, especially in the lungs and pancreas making every breath a struggle and with the pancreas not able to pump out enzymes to break down foods and digest them. They have a lot of trouble gaining weight.
So they can’t cough up all the particles and germs they breathe in every day and they stick in the lungs and grow and become infected. They can’t eat a pizza and gain a pound or two like most others – it’s very hard for them to gain weight. We all need fat on our body to keep our organs healthy and working best they can. We also need fat on our bodies to fall back on when we get sick and can’t or won’t eat.
When and how did you find out that Gwen had Cystic Fibrosis? How was it diagnosed? Where were you?
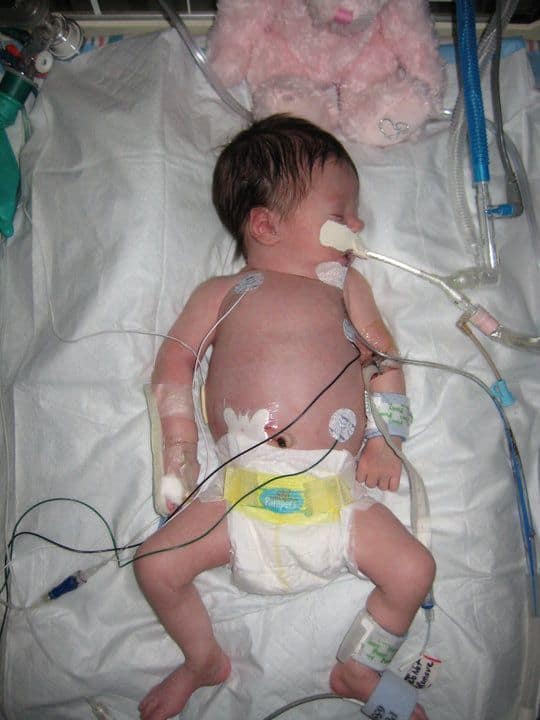 Christopher and I found out we were pregnant in July 2008. We were very excited.
Christopher and I found out we were pregnant in July 2008. We were very excited.
Late February my water broke and off we went to the hospital. Gwen came out via emergency C-Section after her heart rate dropped. She was meconium stained and had to spend her first 9 hours in the nursery to monitor her breathing.
After she got out, we noticed little things, but nothing terribly alarming. She would eat but spit up some after, which happens to the best of babies but this was after every feed. It started becoming more forceful and no longer spit up, but vomit. Her doctor noticed the night before our expected discharge that she had yet to pass her meconium. With the forceful vomiting he decided to transfer us to a nearby NICU.
Once we got there and they gave her a quick once over they told us that they had 3 diagnosis it could be. In order of severity:
- Cystic Fibrosis (Life threatening genetic disease. This was worst case scenario)
- Hirschsprung’s Disease (Intestines missing nerve cells that push poop through your intestines and that part of the intestine is removed)
- Meconium Plug Syndrome (Just can’t pass it on her own and needs a little extra help)
They did a test and ruled out Hirschprungs. After several attempts to clear her of the blockage they decided surgery was necessary. The surgery was performed when she was 9 days old. The surgeon came out after and told me that she has not seen an intestinal blockage that bad except in people with CF and later that day we got confirmation with her DNA/blood test.
She has 2 copies of Delta F-508 which is the “classic” CF gene and presents typically with the worst symptoms.
What were your reactions to finding out that Gwen had Cystic Fibrosis? Can you remember your immediate thoughts?
They pulled us in a room and brought in a social worker and sat down and explained it all to us. It was kind of a blur and I don’t remember much of it except that they told us its unlikely she would make it past 30 years old. That’s really all I remember about that. Its really all I needed to remember about that.
What were the reactions of friends and family about Cystic Fibrosis?
A mixture of understanding and disbelief. My mom realized that her mother lost a sister in infancy to what was recorded as pneumonia but back before Cystic Fibrosis was discovered that’s basically what it came down to. My husbands side spent her first few years saying there is no Cystic Fibrosis on their side and Gwen didn’t get it from him. It’s not always present in family history which is why it was such a shock to us. If we are each 1 out of 25- that’s 24 non-CF carriers we could have ended up with and never had a CF child and never known we even had a Cystic Fibrosis gene.
Had you heard of Cystic Fibrosis before the diagnosis?
I had yes. I remember learning about it in 9th grade science. I remember it was nicknamed “65 roses” because kids couldn’t pronounce their disease correctly but that was pretty much the extent of it.
What were the first few weeks/months of having a new baby with Cystic Fibrosis like?
I can’t really be sure but I would guess it was probably about the same amount of madness for any first time parent.
Gwen was sent home with a NG tube (feeding tube through the nose) and we pulled it out as soon as she got home and got her eating with a bottle. She was never a great eater but she squeaked along for a while. The enzymes we had to give were hard. You have to break open a pill and sprinkle the contents on a spoon of applesauce before every meal. Spoon feeding a 1 month old isn’t the easiest thing in the middle of the night when you’re half asleep and your baby is crying for a bottle.
The worst was the vitamins. They are just awful! Liquid orange vitamins and of course they taste terrible so baby spits them out and they stain everything! Every outfit she had was orange around the collar. It was just a smelly mess.
Aside from all the random Cystic Fibrosis medications, the rest was pretty easy to deal with. We had to do manual chest percussions with little plastic cups and give Nebulized Albuterol twice a day. She would sleep through that every time which made it easier to do than fight a baby as you pat them on the chest hard for 20 minutes. It was a heck of a lot of info to take in at once and once you are able to get past the shock that you will undoubtedly live past your child, its just a matter of routine. New parents all have dishes and laundry and stuff, we just had more to do on top of that with giving/organizing meds, cleaning nebulizers etc.

What was the advice you were given/told about the disease? Treatments, medications, life expectancy…
We really weren’t given much. Basically the NICU doctor told us it’s a terrible disease and she will suffer for her short life. They had only ever seen 1 other kid go through there with Cystic Fibrosis and were basing what they told me, off what they all learned years/decades prior in medical school.
When we started her at the Cystic Fibrosis clinic at Childrens Hospital of Los Angeles, they were a lot more up to date and positive about her outcome. They told us if we can get her through her first 4-5 years without any major illness it will add years to her life. We put germs on our hit list and we kept her from most all other kids and out of day care, play areas and parks and for the most part, it did its job. She got her first real illness at 5 years old.
Did having a second child worry you? Did you know Gena might/might not have Cystic Fibrosis too?
From the beginning of my pregnancy I just felt like she was fine. I was convinced she didn’t have Cystic Fibrosis – I could just feel it. Well intuition not being a total failure on my part, our second child has been exceptionally healthy even though she does have Cystic Fibrosis. Her weight is great, as well as her lungs. She’s doing beautifully!

When/how did you find out that Gena had Cystic Fibrosis? How was it diagnosed? Where were you?
We had a planned C-section with her because of Gwen’s delivery. They wanted to schedule delivery early so that Gena, if she did have Cystic Fibrosis, would have a better chance at passing her meconium since it gets thicker the longer shes in. So they took her out at 39 weeks and everything was fine. She passed her meconium but the doctor noted that it was “jelly like” and sent for a blood test while we were still in the hospital. We got the results a few days after we got home and yes she was positive.
What were your reactions to finding out your 2nd daughter had Cystic Fibrosis? Can you remember your immediate thoughts?
Obviously I would never wish Cystic Fibrosis on anyone, especially not my child, but I remember thinking before we got the results, that if she was positive for CF, that the silver lining would be at least her and Gwen can go through that together. I watch my daughters with this disease every day, but I will never know what its like to have the disease and they will have that with each other.
I’m not sure how common knowledge it is among non-CF families but CF people are not supposed to be around each other. They can spread germs and illnesses that the other person might not be able to fight off and it could be fatal. So in my eyes having that close friend that really does understand your disease because they have it to is something positive to come out of such a negative situation.
How do your daughters differ with their Cystic Fibrosis?
A lot! Gwen has the common typical Cystic Fibrosis symptoms. Terrible weight gain and some lung issues where as Gena really has no issues (so far). It wont last I’m sure though as Cystic Fibrosis is a progressive disease.
What does a typical day in the life of Cystic Fibrosis parent look like?
Similar to what every parent goes through except we have a few added steps. Lung treatments, meds, extra doctors appointments and time in the hospital. That’s the hard part – trying to figure out what to do with one kid while the other in is the hospital and how to split your time between them.
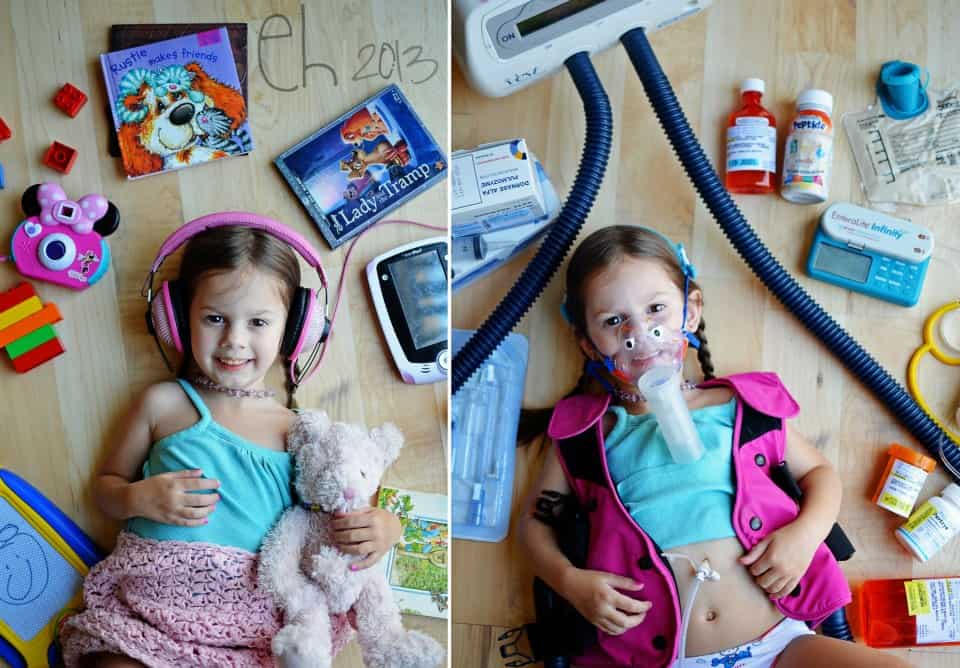
Are the girls aware/know/understand that they have CF? How do you think that they cope?
Gwen knows she has it. We are honest with her about it. We havn’t exactly told her about her shortened life. We try and keep it age appropriate best we can.
Gena just knows that her and her sister take pills. I don’t think she realizes yet that other kids don’t do that.

What medications/how much medication do they have to take?
That changes. Currently they are on the least amount of medications they have been on yet. Gwen takes 3 enzyme pills, every 3 hours, round the clock while on her 24 hour tube feed. She takes a Multi Vitamin, Prevacid, and Symbicort 2 times a day and with her Symbicort she does VEST treatments 2 times a day for 20 minutes each. The VEST uses technology called High Frequency Chest Wall Oscillation (HFCWO). During therapy, the inflatable garment inflates and deflates rapidly, applying gentle pressure to the chest wall. This works to loosen and thin mucus and to move it toward the larger airways, where it can be cleared by coughing or suctioning.
Genevieve is on the same except instead of Symbicort, she takes Albuterol 2 times a day.


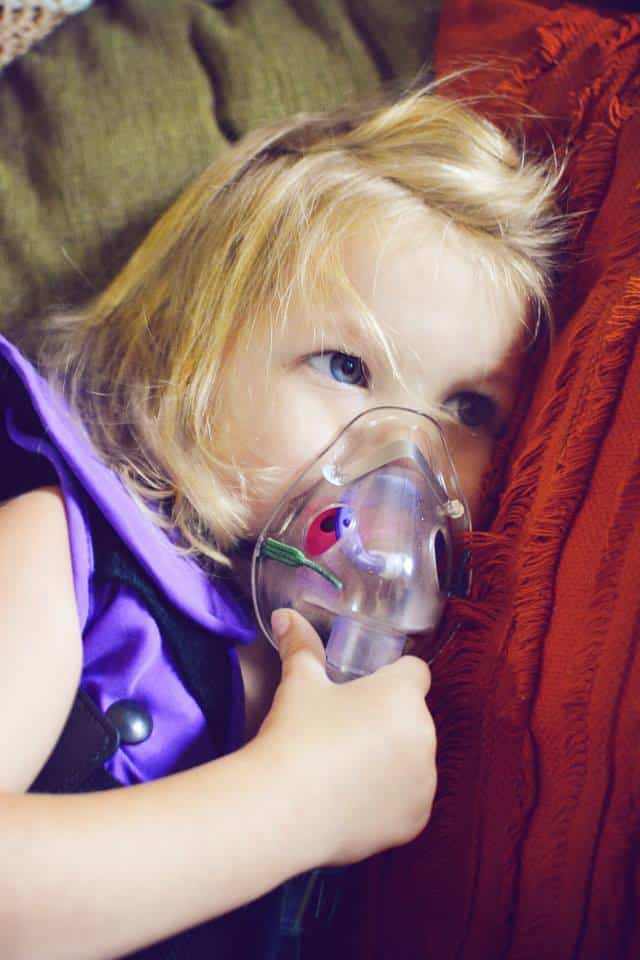

What are your hopes and dreams for the girls?
That they live long happy. It’s all any parent wants.
What are the girls’ hopes and dreams?
Gwen is full of dreams. She has many different careers and goals in life. She wants to be in a band, ride motorcycles, race cars, dance ballet and make video games like her dad.
I think Gena hopes to own a world supply of binkies!


Is there anything they can’t do, that a “normal” kid would do?
Gwen is currently limited at school because she is on 24 hour tube feeds and the school won’t let her play at recess for fear her tube getting caught on something. She does tire quicker than other kids and loses more salt and fluids so we have to keep an eye on her on hot days or excessive physical activities. Aside from that she can pretty much do it all.
As an adult though, their job options are not as open as healthy folks. They’re probably not going to be on NASA’s top pick list and probably not the best idea to be working around hospitals and such, but kids with Cystic Fibrosis are living long enough now that they can go on to college and think about starting careers which hasn’t happened until recently with medical advances.
What has your girls’ Cystic Fibrosis taught you?
Every day counts!


What do you think is the most important thing about raising a child with Cystic Fibrosis?
To each their own and all, but I’ve seen too many kids try and hide their differences and the damage it can cause. That’s why I am 100% open with the girls about their disease. Its not something they should hide. It’s a part of them and who they are. It needs to be accepted for their sake and for the sake of their health.
The rest comes down to how you want to treat their disease. Some people just treat them like every other kid – live their life as full as they can, for as long as they got. Then others put their kids in bubbles and try and pack on every extra minute they can on to their life.
I’ve been trying to balance both.
We take her out of class to go camping, but we also make sure she’s going to class and learning for her future. Keeping her away from sick kids, but letting her play and socialize.
As a mother it’s hard to know in your head you aren’t doing everything you can to keep your child alive as long as they possibly can, but if it were me, I would want to be out living life and enjoying the time I have. It’s a personal battle I have to deal with daily.

What advice would you give to parents who have just had a child diagnosed with Cystic Fibrosis?
I don’t really have much advice, but the one thing that helped me along in accepting the diagnosis is the poem Welcome to Holland by Emily Perl Kingsley.
Tell me about Hope for Hughes and Great Strides…
We used to be called Team Gwen but when Genevieve was born, we switched our team name to Hope for Hughes.
Every year we participate in the Cystic Fibrosis Foundation’s Great Strides. It’s an annual walk to raise money and awareness for Cystic Fibrosis, in our effort to find a cure for our girls and the 70,000 people world wide that have CF.

Life expectancy has shot up a lot in recent years from advancements in medicines for Cystic Fibrosis, but for a child born right now, 37 years is still not nearly enough. I’m 32 and I would not be done with my life in 5 years. My kids would not be ready for me to go in 5. It’s just not enough.
Cystic Fibrosis is the #1 genetic killer of children today in the US and that needs to change. This disease needs a cure but we can’t do that without the help of others. Money for research and awareness are our two best weapons in our fight against Cystic Fibrosis.
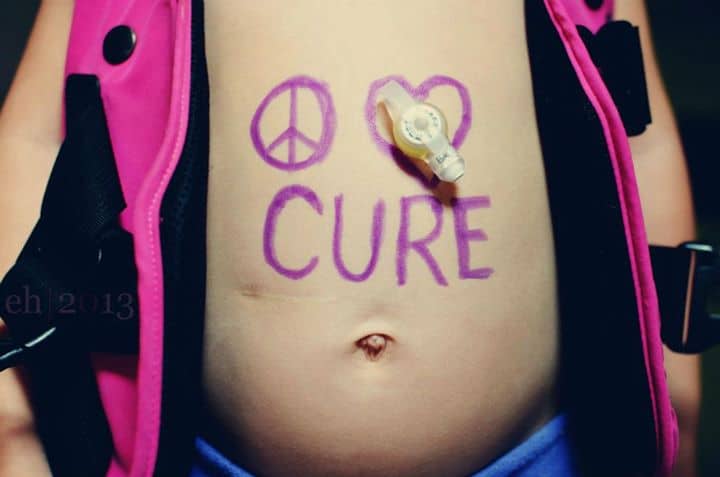
I want to thank my friend Elisa for answering my questions about Cystic Fibrosis, and for helping me (and I hope you too) understand more about the disease and what the life of a mother of children with CF is like.
I went to school with a boy who had Cystic Fibrosis. He died shortly after his 16th birthday. When I first “met” Elisa and she told me her daughter had CF, I mentioned about him, as I didn’t really know much about the illness. I remember her saying how treatments have come a long way, but more still needs to be done.
I always hope that one day, CF really will stand for Cure Found. If you wish to donate to help the cause, then you can do so in the US through Great Strides, or in the UK through the Cystic Fibrosis Trust.
It also helps if you share to help raise awareness about CF, so I know that Elisa and her family would appreciate this post being shared far and wide – please consider hitting the share buttons at the bottom of this post. Thank you. ♥

Arwen
September 5, 2021 @ 7:30 am
I don’t know the best way to put this, but those girls are my cousins! And I’ve only met them maybe twice, but they were very sweet, and fun to play with. At first I was very scared of touching them, because my mom had told me they could get sick, and at the end of our family reunion Gwen came up and gave me a hug. I was terrified ( not of me getting sick, obviously ).
But when I told my mom she said that it was okay, because their mom wanted them to have a happy life, where they didn’t have to be too scared of anything. I don’t know that part of my moms side of the family very well, because they live quite far from us, but they were very nice. And recently ( as in 2019 recently ) a treatment was developed for those with CF. I’m very glad for that. And glad that you have this page. Have a wonderful day and god bless!
Michelle
September 5, 2021 @ 11:33 am
Wow, what a small world! I think that the powers that be are getting closer to finding a cure, and I hope that it will save all those with CF!
Ickle Pickle
May 27, 2015 @ 8:56 pm
What an incredible post. So moving, what a great family. Love all the photos . Kaz x
Michelle Ordever
May 28, 2015 @ 9:18 am
Thank you for reading xx
Stephs Two Girls
May 20, 2015 @ 10:31 pm
They are beautiful girls. Thanks for sharing, I have learnt more about CF than I knew before x
Michelle Ordever
May 22, 2015 @ 8:52 am
Thank you for sharing this over on Twitter xx I learned a lot through writing this up x
Vaichin@RamblingThroughParenth
May 18, 2015 @ 4:36 pm
What a truly inspirational family! ‘Every day counts’ is a message we could all live by.
Michelle Ordever
May 22, 2015 @ 8:52 am
I completely agree x
Helen
May 18, 2015 @ 2:54 pm
Great piece of writing which explains more about CF and raises awareness. Thank you for posting.
Michelle Ordever
May 22, 2015 @ 8:52 am
Thank you for reading, am glad it brings awareness, which I know is what Elisa wanted
Carolynne @ Mummy Endeavours
May 17, 2015 @ 11:11 pm
What beautiful little girls. Sounds like the mum is doing a great job. Such a horrible disease that I didn’t know that much about. Thanks for sharing and fantastic to be raising awareness and funds
Michelle Ordever
May 22, 2015 @ 8:53 am
Thank you very much for reading. She and her husband do an amazing job!
Jaime Oliver
May 16, 2015 @ 11:22 pm
what a really informative piece, thank you to your friend for being so open, the more people that know the more people will support :-)
Michelle Ordever
May 22, 2015 @ 8:53 am
Thank you for reading x
Globalmouse
May 15, 2015 @ 1:56 pm
This is great that you’re raising so much awareness and helping out as much as you do. This was really interesting and hard to read, thank you for sharing.
Michelle Ordever
May 15, 2015 @ 6:58 pm
I always say to my friend that I’m never really sure how much I’m doing actually helps, but she assured me that just the awareness is enough to help. It was a hard read for me when she emailed me back, I mean, I knew the girls have CF, but everything that goes with it from my friends mouth… it made me cry
Beautyqueenuk
May 14, 2015 @ 10:04 pm
I really do hope there is a cure found, what beautiful happy ones they both look x
Michelle Ordever
May 15, 2015 @ 6:57 pm
They are such lovely little girls! x
Kara
May 14, 2015 @ 11:00 am
My husband lost his childhood friend to this disease. I hope a cure is found soon
Michelle Ordever
May 15, 2015 @ 6:57 pm
Very sad… I believe the life expectancy is a lot higher these days x
Mama Syder
May 14, 2015 @ 8:57 am
Such a beautiful post, really informative. Hope a cure is found soon x
Michelle Ordever
May 15, 2015 @ 6:56 pm
Thank you – me too x
pinkoddy
May 13, 2015 @ 11:42 pm
I hope they do find cure. So beautiful that they are so happy. Great way of sharing the information too.
Michelle Ordever
May 15, 2015 @ 6:56 pm
Same here… thank you.
Jen Walshaw
May 13, 2015 @ 10:08 pm
CF is such a horrible life limiting disease. We lived for nearly a year thinking that Maxi had it! My next door neighbors 12 year old is a sufferer
Michelle Ordever
May 15, 2015 @ 6:56 pm
Wow, that must have been very hard for that year :( Let’s hope they find a cure one day
emmaand3
May 13, 2015 @ 8:20 pm
Such a positive and powerful post.
Michelle Ordever
May 15, 2015 @ 6:56 pm
Thank you x
angela
May 13, 2015 @ 2:51 pm
I really hope they find a cure soon. Thank you for raising awareness xxxxx
Michelle Ordever
May 15, 2015 @ 6:55 pm
Me too – for their sakes xx
Erica Price
May 13, 2015 @ 1:13 pm
There has to be a way doesn’t there? Hope they find it soon. I can’t imagine living with the knowledge that my child has a shortened lifespan – it must take a lot of strength.
Michelle Ordever
May 15, 2015 @ 6:55 pm
I hope so – for this, and other life limiting diseases.
Happy Homebird
May 13, 2015 @ 12:56 pm
I felt very humbled reading this, what beautiful little girls and how much they have to go through. Brave children and parents, I did not know a lot about CF before reading this. Thank you for telling this families’ story x
Michelle Ordever
May 15, 2015 @ 6:55 pm
Same here. I’ve just seen that they’re heading off to go camping after an appointment at the hospital, so to me that is living life to the absolute fullest! x
Bek B
May 13, 2015 @ 12:37 pm
Wow-what an informative post. Thank you for the amount of detail, I feel like I have learned a lot from this. I had a friend whose sister had CF but never fully understood what that meant. What lovely girls and fantastic parents.
Michelle Ordever
May 15, 2015 @ 6:54 pm
I learned a lot too – I felt with me doing the month of purple in May for my photos, I needed to find out even more about the disease.
Sarah Ebner
May 13, 2015 @ 11:26 am
This is so moving and also inspirational. What wonderful little girls (and so gorgeous too) who are lucky to have such terrific parents.
Michelle Ordever
May 15, 2015 @ 6:54 pm
Just reading my friends words moved and inspired me too. They are gorgeous aren’t they!